Theory of Molecular Manipulation
Using density functional theory and molecular mechanics simulations, we study the properties of metastable molecular conformations created by single-molecule manipulation with scanning probe microscopes (SPM).
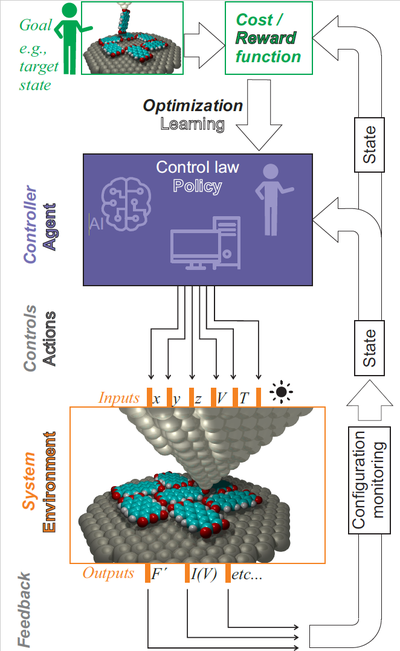
Molecular manipulation with an SPM [1, 2] leads to tip-molecule-surface junctions with a rich variety of conformations that are, however, not directly observable. To understand the properties of these junctions and thereby to advance proactive molecular fabrication, we analyse our experiments with the help of molecular mechanics simulations [3, 4, 5] and density functional theory calculations. This helps us to explore the stabilisation mechanisms behind metastable molecule-surface conformations. Van der Waals forces [4] and electrostatic interactions [6, 7], which both act over a long range, are of particular importance for the correct description of such metastable conformations.
A central goal of our research is establishing the observability of molecular conformations during the manipulation experiment. Here, density functional calculations allow linking experimentally accessible junction properties (stiffness, electrical conductance) to actual atomically precise molecular conformations. On this basis, the inverse problem can be solved, for example by means of control theory.