Polymer structure and dynamics
The rheological properties of many polymer systems are of great relevance in terms of their application and industrial processing. These are determined by the interaction and movement of the constituent structural units. Understanding this process is indeed a challenge with the aim of facilitating the molecular design of new materials.
The dynamic spectrum of polymers is very elaborate, and is currently described in terms of entropy-driven Rouse dynamics along with the reptation model involving creeping movements along a tube formed by the mutually interpenetrating chains. This tube model is typically described using a mean field model; macroscopic properties are thus linked to the molecular and microscopic aspects of the structure and dynamics of polymers of varying architecture.
Branched Polymers
In linear polymers the microscopic origin of elastic and flow properties is related to their mutual entangling and their relative motion. Different relaxation processes can be identified and assigned to different structural details.
The embedding of chains in their surroundings naturally introduces the concept of tube-confinement and in connection with this, a set of well-defined molecular timescales. The relaxation time spectrum is severely slowed down and modified by the introduction of knots or branching points and gives rise to a hierarchical development of relaxation in dependence of the architectural complexity. Blends of linear and branched polymer chains combine their respective properties and lead to improved processing stability compared to the simple chain.
Our actual research program deals with H-shaped and 2nd generation dendrimer-like systems in linear matrices in the non-linear viscoelastic region. Relaxing structures are investigated by a controlled quench below the glass transition temperatures and subsequent relaxation phases above Tg in situ in the neutron beam.
Ring Polymers
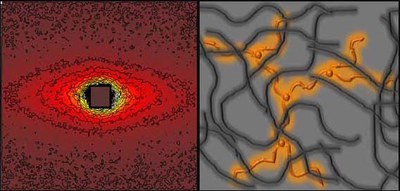
Ring polymers constitute a special class of architectural polymers. Their structure and linear viscoelastic response can be investigated nowadays in great detail due to their availability in sufficiently large quantities thanks to a new chromatographic technique with extreme topological sensitivity.
The closed structure gives rise to new and non-universal aspects of the description of polymer dynamics, as no tube-like entanglement can exist and any kind of chain relaxation involving chain ends is absent.
Microscopic studies of the structure and dynamics of ideal ring polymers of various chain lengths in both pure and blended states with linear matrices are performed and supported by the appropriate macroscopic techniques such as rheology or dielectric spectroscopy. The observed relaxation behaviour during experiments resembles that of lattice animals or randomly-branched polymers.
As the rings do not form entanglements, they are an ideal probe to sample the environment. In this way, dynamic aspects and movement mechanisms in polymer melts and solutions can be sampled with great sensitivity.
Polymer Networks
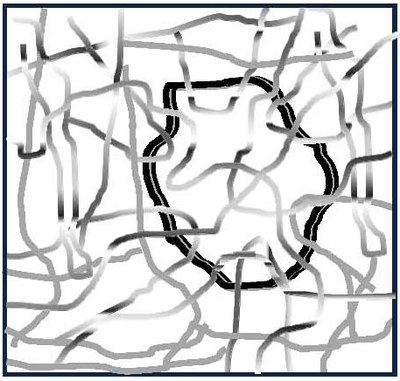
The now well-established tube concepts for entangled melts, historically developed for rubber polymer networks, ensured topology conservation by assuming that the polymer chains are not allowed to cross, and segment fluctuations are restricted in a potential. Their dynamical short time behaviour, determined by in-tube dynamics, is very similar in this case. The stress-strain behaviour of permanently cross-linked rubber under experimental conditions, however, displays the contributions of both crosslink and entangling states.
In non-affine tube model theories, the span of fluctuations around the tube axis in densely-entangled networks is linked non-linearly to macroscopic deformation. Research on the molecular description of deformed tube diameters and the chain deformation in model systems currently focuses on the combined use of static and dynamic neutron scattering techniques in model networks.
Newly developed networks, which have attained a high degree of adaption by means of permanent networking and transient links, are a new challenge. In order to clarify these mechanisms, the length and time scales recorded by neutron scattering are essential.
Tube Models

The elastic behaviour of polymers is strongly related to microscopic confinement length i.e. tube diameter. The existence of this single particular length scale, however, has been called into question. The hypothesis of two length scales, one longitudinal and one perpendicular to the primitive path is, on the other hand, not proven either.
Furthermore, the nature of entanglement may depend on the architectural complexity involved. If the tube dimensions vary in size, both sizes may be differentiated by altering the stiffness of the polymer chain. Then again, discrete entanglements as opposed to a continuous tube picture may shed light on the intrinsic nature of the chain topology.
These studies are performed on syndio-tactic polypropylenes and mixtures of ring and linear polystyrene polymers; linears intermingle but protrude out from the cyclics, leading to more localized entangling states. Neutron scattering experiments are complemented here by macroscopic rheology and dielectric spectroscopy methods.
Self-healing and Supramolecular Associations
Bio-inspired hydrogen bonding is the prominent key factor for the design of novel self-healing properties. These counteract damage processes directly and autonomously on the molecular level. The mechanism is complex and multi-scaled.
A microscopic and physical understanding of the self-healing process is therefore only feasible through a combination of neutron scattering and complementary laboratory macroscopic techniques, such as rheology, dielectric spectroscopy and pulse field gradient nuclear magnetic resonance. An understanding of the self-healing process allows generalizations to be drawn and will eventually lead to the controlled design of new adaptive high-performance composites.
The hydrogen-bonding mechanism may be specifically implemented in a network, so that in addition to the usual covalent chemical cross linking, a transient but reversible link fraction is also present. As a result, a catastrophic failure in materials through crack propagation, for example, can be prevented by the self-healing of molecular cracks from the very beginning, i.e. on a molecular level. Furthermore, the stress-relieving dissipative source reduces wear and tear and prolongs the lifetime of such materials.
The supramolecular structural formation is based on a complex interplay of polymer building blocks and (self-) complementary groups. Studies on transient branched or cross-linked polymers and networks open up new research areas for knowledge-based material development. With the help of transient branches, for example, in the formation of chains with a comb structure, the processing properties can be selectively altered or new equilibrium properties specifically adjusted.
Grafted Polymers
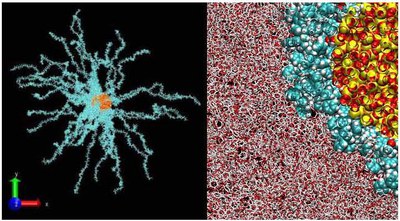
Nanometer-sized fillers result in an enormous increase of interface area and polymers reinforced with nanoscale particles are therefore expected to show improved properties. The multi-functional behaviour of new nanocomposites, where the decrease in viscosity is particularly advantageous for processing, is related to particle/matrix interactions in the interphase between the bulk and the surface chemistry.
Model systems that allow perfect dispersion via the grafting of monodisperse polymer arms onto the surface area are currently being investigated. The focus is placed on their structure, dispersion and the density profile in matrices with varied levels of entanglement.
The scattering experiments are supported by complementary physical methods. By fully utilizing the contrast variation, the focus can be placed either on the particle, the integral grafted shell or selectively on the matrix polymer conformation.
Polymers Under Confinement
Understanding the structure and dynamics of confined polymers is important for applications in nanotechnology such as coatings for electrical devices, lubricants, and polymer nanocomposite materials. At present, a molecular theory which predicts the properties of the composite based only on the ingredients does not exist. Therefore, the ultimate vision is to derive the macroscopic properties from the microscopic parameters.
In order to achieve this goal, we study the influence of surfaces and confinement on the dynamics of polymer melt using neutron scattering methods. Our focus is on 3D confinements (Fig.1a) presented by SiO2 nanoparticles and 2D confinements such as cylindrical nanopores of anodic aluminium oxide or silica (Fig.1b). Neutron scattering techniques offer the unique possibility to observe single polymer chain conformation and dynamics in these systems.
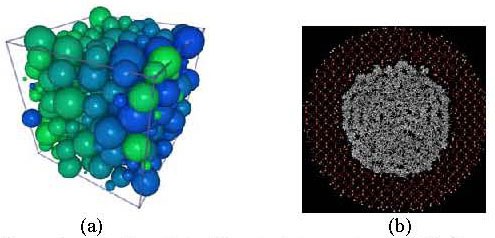
(a) 3D confinement presented by nanoparticles (b) MD simulations of a polymer confined in silica nanopores (2D confinement).
Dynamics in Soft Confined Morphologies
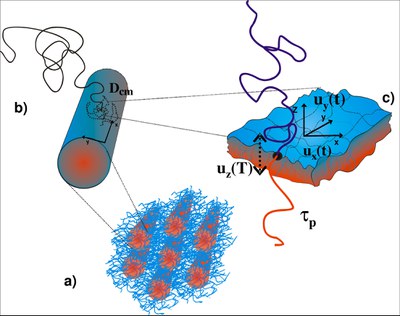
Block copolymers with thermodynamically incompatible blocks self-assemble into various microstructures including spherical, cylindrical, lamellar or bicontinuous mesophases depending on the block compositon.
One of the open questions is how the polymer dynamics is affected in the nano-structured domains of a segregated diblock copolymer system. Due to the limited space within the domains and the presence of a junction zone, a specific confinement effect is to be expected. Studies are performed using neutron spin echo spectroscopy combined with sophisticated hydrogen/deuterium labelling to individually highlight single chain dynamics and the dynamics of the interface.
A sketch of the dynamics in a polyisoprene (PI)-polydimethylsiloxane block copolymer melt forming cylindrical PI domains is shown in the above figure. Possible processes are (a) the centre of mass motion of individual cylinders, (b) single chain diffusion along the interface of the cylinder, and (c) local undulations of the interface. Further studies will be performed on block copolymers and the forming of spherical domains.
Kinetics in Block Copolymer Micelles
We investigate kinetic processes in block copolymer micelles. Thereby we distinguish between chain exchange kinetics, formation kinetics, and shape transition kinetics.
Chain exchange between different micelles is an inherent property at thermodynamical equilibrium. Time scales and underlying mechanisms are investigated. Formation kinetics is concerned with the route by which amphiphilic block copolymers self-assemble into micellar structures.
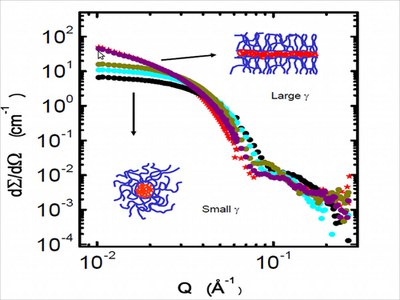
Studies of transition kinetics yield information on the reorganization mechanism during the transition between different micellar geomtries, e.g. from cylinders to spheres (see figure). The kinetics are accessible by monitoring structural changes with time-resolved small angle x-ray and neutron scattering experiments.
In order to observe the chain exchange kinetics, a particular hydrogen/deuterium contrast variation scheme has been developed. The general aim of the kinetic studies is to fundamentally understand the mechanisms of self-assembly which is a common building principle seen throughout nature and technology.